Nanostructure
Akio Ishii, Nobutomo Nakamura,“Parameter-free multiscale analysis of hydrogen solubility in Pd nanofilms under hydrogen gas using density functional theory”, Journal of Applied Physics, 137 (2025) 064305.

A parameter-free multiscale analysis of hydrogen solubility, hydrogen coverage, and hydrogen bulk concentration for face-centered-cubic Pd nanofilms with a (111) surface under hydrogen gas conditions is proposed using density functional theory and a simple kinetic model. The calculated solubility is quantitatively comparable to that obtained via experimental observations. Although the Pd surface is fully covered by hydrogen in a short time (microseconds to milliseconds) under exposure to 10–10000 ppm hydrogen gas pressure (1.0 atm), the hydrogen concentration in the subsurface or bulk changes significantly on the experimental time scale depending on gas pressure. We confirmed that the hydrogen concentration in the bulk or subsurface of Pd nanofilms (not the Pd surface) plays a role in the resistance change of Pd through comparison between the calculated hydrogen concentration and experimental observations of an electric resistance change caused by exposure to hydrogen gas. A hydrogen sensor requires a 0.1% change in the hydrogen concentration in the bulk to observe a significant change in the electric resistance. Furthermore, we calculated the time-dependent diffusion coefficient of hydrogen in a Pd nanofilm and compared it with the experimental observed one. We also investigated temperature dependency of the solubility and confirmed that the hydrogen gas pressure determines hydrogen solubility in Pd nanofilms at the equilibrium state, whereas the temperature controls the speed to reach the equilibrium state.
Back to topShihao Zhang, Shigenobu Ogata,“Dislocation plasticity in c-axis nanopillar compression of wurtzite ceramics: A study using neural network potentials”, Journal of the American Ceramic Society, e20406, (2025) 1-11.
Ceramics typically exhibit brittle characteristics at room temperature. However, under high compressive stress conditions, such as during nanoindentation orcompression tests on nanopillars, these materials can reach the necessary shear stress for dislocation nucleation and glide before fracture occurs. This allows for the observation of plastic deformation through dislocations even at room temperature. Yet, detailed insights into their atomic-scale dislocation plasticity remain scarce. This study employs atomistic simulations to explore the dislocation plasticity in c-oriented nanopillars of wurtzite ceramics (i.e., GaN and ZnO) under uniaxial [0001] compression, utilizing a specially developed first-principles neural network interatomic potential. We observed activation of {0111}⟨1123⟩ and {1122}⟨1123⟩ dislocations, corroborating experimental findings from insitu compression tests. Moreover, a wurtzite-to-hexagonal phase transformation begins at facet edges and extends inward, forming wedge-shaped regions. As compression progresses, dislocations nucleate at the wurtzite-hexagonal phase boundary and spread throughout the nanopillar, contributing to a rougher surface texture. These quantitative results offer new insights into the plastic deformation behaviors of nanopillars under compressive loading, highlighting the potential of dislocation engineering in these ceramics.
Back to topAkio Ishii,“Edge- and vertex-originated differences between nanoparticles and nanovoids: A density functional theory study of face-centered-cubic Al”, Computational Materials Science, 246 (2025) 113342.

The differences between nanoparticles and nanovoids cannot be clearly distinguished energetically using conventional comparisons based on the surface energies of these species. For example, nanoparticles and nanovoids with the same volume and shape are considered energetically equivalent to the conventional Wulff construction, and so the difference in their morphology cannot be evaluated. This can be attributed to fact that using such approaches, the effects of excess defects, edges, and vertices in nanoparticles and nanovoids are typically ignored. In this study, we investigated the energetic differences between face-centered-cubic (FCC) nanoparticles of Al and nanovoids in bulk FCC Al structure with conventional truncated octahedral shapes by calculating the excess energies attributed to their edges, vertices, and sizes. This was achieved using density functional theory calculations and our previously reported method for evaluating the effects of edges and vertices. The morphological differences between the nanoparticles and nanovoids were also discussed based on the obtained results.
Back to topNobutomo Nakamura, Kazushi Yoshikawa, Akio Ishii,“Enhancement of hydrogen response by forming an Au submonolayer on nanogap Pd nanoparticles”, Applied Physics Letters, 125 (2024) 021902.

When Pd nanoparticles dispersed on a glass substrate with nanometer order gaps are exposed to H2 gas, H atoms are adsorbed on the nanoparticle surface, and the electrical resistance between the nanoparticles increases because of the tunneling current suppression. In contrast, when Au nanoparticles are exposed to H2 gas, the resistance remains unchanged because H atoms are not adsorbed on the Au surface. Considering these behaviors, the change ratio of the electrical resistance is expected to be smaller when the surface of Pd nanoparticles is partially covered with Au. However, the experimental results show the opposite resistance change. Density functional theory simulation indicates that H atoms are adsorbed and absorbed on the pure Pd surface, but H atoms are adsorbed and tend to remain on the partially covered Pd surface. These results indicate that the decrease in the resistance due to the gap narrowing by hydrogen absorption occurs in Pd nanoparticles, but it does not occur in Au/Pd nanoparticles, resulting in a larger resistivity increase compared with the Pd nanoparticles. This result implies that in certain cases, the low reactivity of Au to H2 contributes to the enhancement of the electrical resistance response.
Back to topAkio Ishii,“Energetical effects of the edges and vertices of face-centered-cubic Pd and Au nanoparticles: A density functional theory study”, Computational Materials Science, 243 (2024) 113122.

The properties of nanoparticles depend on their sizes, and these size effects in face-centered-cubic (FCC) nanoparticles are attributed to the edge and vertex effects. However, the effects of edges and vertices on the properties of nanoparticles have not yet been explicitly investigated. In this study, we propose a method to evaluate the edge and vertex effects in FCC nanoparticles using density functional theory atomistic simulations. Pd and Au FCC nanoparticles are modeled as conventional truncated octahedra with {111} and {100} faces. The changes in the excess energy due to the edges and vertices are separately described and are calculated with respect to the size of the nanoparticles. Through explicit calculations, we confirmed that for Pd and Au nanoparticles with several hundred atoms, the vertex effects are negligible, whereas the edge effects are still significant.
Back to topGang Wu, Shigenobu Ogata and Lei Gao,“Atomistic simulations of the frictional properties of 2D materials: a review”, Journal of Physics D: Applied Physics, 57 (2024) 293001.
The two-dimensional (2D) materials are regarded as the ideal solid lubricants at micro- and nano-scale. Besides the experiments and analytical models, the atomistic simulations are important tools to investigate the frictional properties of 2D materials. This review will focus the recent atomistic simulation studies on frictional properties 2D materials with a particular emphasis on the density functional theory (DFT) calculations and molecular dynamics (MD) simulations. Starting from the proper calculation of long range dispersion forces, the correlations between the physical characteristics (e.g. electronic charge redistribution, interfacial commensurability, chemical modification, moiré superlattice, layer effect, atomic contact quality, defect, external fields, humidity and temperature) and frictional properties of 2D materials are reviewed for both the interlayer and surface sliding. Meanwhile, recent MD simulations about the phononic energy dissipation in friction of 2D materials are summarized. At last, some shortcomings in current simulation techniques are summarized and it is suggested that the atomistic simulations combined with machine learning will be a more powerful strategy to investigate the frictional properties of 2D materials.
Back to topAkio Ishii, Nobutomo Nakamura,“Ab initio morphology prediction of Pd, Ag, Au, and Pt nanoparticles on (0001) sapphire substrates”, Journal of Applied Physics, 135 (2024) 094301.
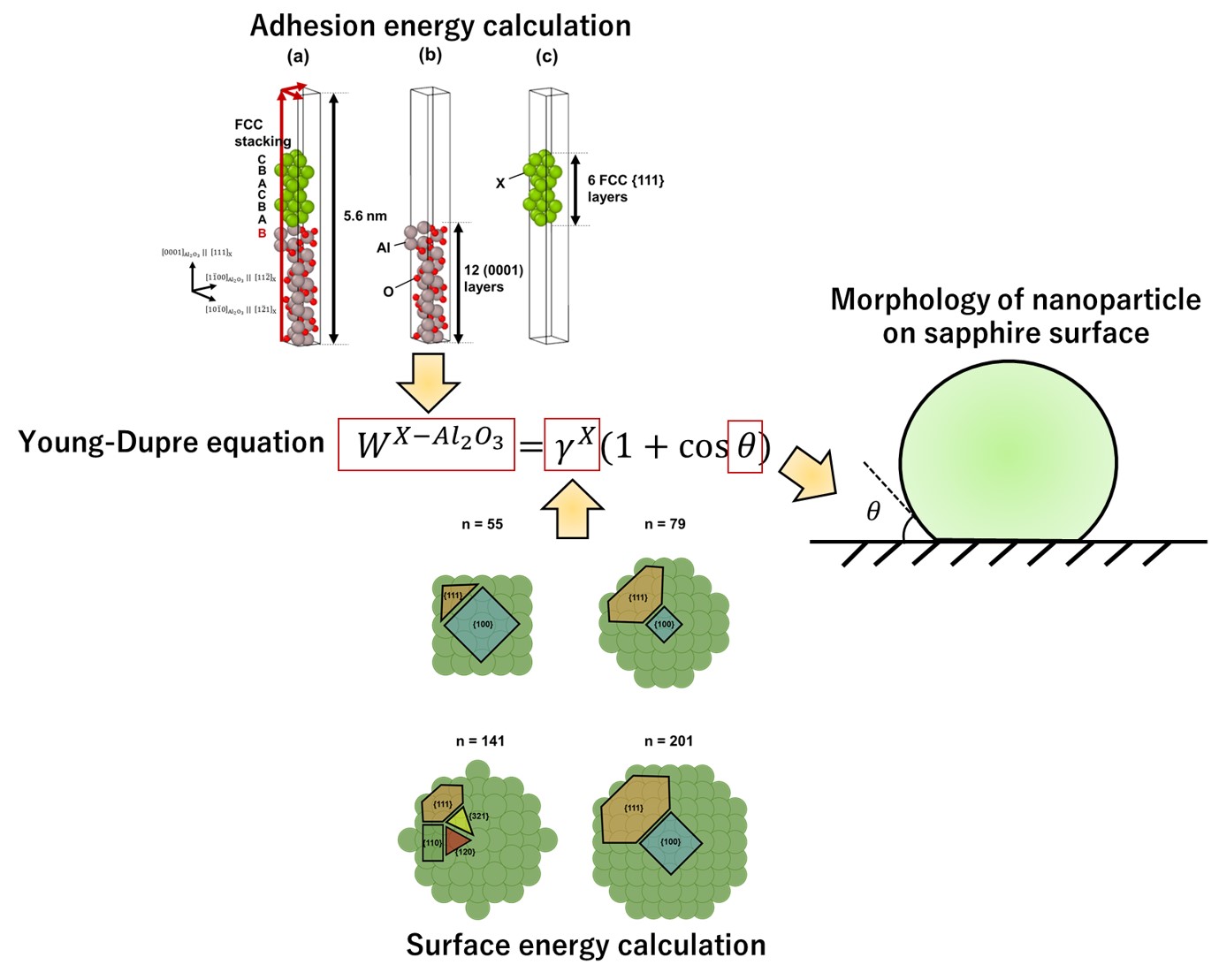
We energetically predict the morphology of Pd, Ag, Au, and Pt nanoparticles on (0001) sapphire substrates, using density functional theory (DFT) simulations and the well-known Young–Dupre equation. In all cases, the contact angles exceed 90° , indicating that the nanoparticles are spherical. Notably, Au nanoparticles exhibit a higher contact angle than those of their counterparts. The validity of the proposed abinitio nanoparticle morphology prediction approach based on DFT simulations was assessed in comparison with our previous experimental findings pertaining to the time variation of the full width at half maximum (FWHM) of the resonant peak. Furthermore, the diffusivities of single Pd, Ag, Au, and Pt atoms on the substrate were evaluated by calculating the activation energy, offering insights into the underlying physics governing the timing of FWHM peaks. The analysis confirms a higher diffusivity of Au and Ag compared with Pd and Pt. According to the comparison between DFT and experiment results, although no clear relation is observed between the contact angles and timing of FWHM peaks, the diffusivity of sputtered atoms may influence the timing of FWHM peaks. Thus, timing can help to clarify the nanoparticle size, rather than shape.
Back to topT.L. Dora, Sandeep Kumar Singh, Radha Raman Mishra, Eric R. Homer, Shigenobu Ogata, Akarsh Verma,“Deformation and boundary motion analysis of a faceted twin grain boundary”, International Journal of Mechanical Sciences, 269 (2024) 109044-1-11.
In this article, molecular dynamics simulations are used to understand how a nickel bicrystal with faceted incoherent Σ3 grain boundaries respond to uniaxial tensile loading. The deformation response is studied over a wide range of temperatures (100 – 900 K) and strain rates (107 – 1010 s−1). The dislocation extraction algorithm and common neighbor analysis are employed to identify the deformation mechanisms. Our results reveal that the yield stress decreases with temperature and increases with strain rate; whereas the elastic modulus decreases with temperature and is independent of strain rate. Furthermore, incipient plasticity is detected ahead of the yield point at lower temperatures and lower strain rates. Interestingly, the incoherent twin grain boundaries are quite mobile under the uniaxial tensile loading at lower temperatures and lower strain rates. But this mobility decreased at higher temperatures and higher strain rates, thereby, confirming this faceted grain boundary's non-Arrhenius (anti-thermal) migration behavior even under mechanical loading. From a deformation perspective, the incoherent twin facet of the grain boundary served as the major source for stacking fault formation at lower temperatures and higher strain rates. However, with the increase in temperature, the stacking faults became shorter and originated from both the incoherent twin facet and the tips of coherent twin facet. These results are in qualitative agreement with the experimental results documented in the literature.
Back to topChih-Jen Yeh, Chang-Wei Huang, Yu-Chieh Lo, Shigenobu Ogata, Ding Yuan Li, Hsuan-Teh Hu, Jason Shian-Ching Jang,“Effect of nanoglass grain size investigated by a mesoscale variable characteristic strain model”, International Journal of Mechanical Sciences, 266 (2024) 108981-1-15.

Severe shear localization in metallic glasses (MGs) significantly limits their mechanical performance. Nanoglass (NG), composed of heterogeneous glassy domains created by introducing interfaces into MGs at the nanoscale, could be a promising strategy against severe shear localization, as demonstrated by numerous atomistic simulations. This study introduces a novel mesoscale kinetic Monte Carlo (kMC) model with a variable characteristic strain (VCS) to investigate the grain size effect in NGs. This model captures the complex evolution of shear bands during deformation, revealing a surprising transition from inhomogeneous to homogeneous deformation as the NG grain size decreases to approximately 10 nm. This transition is attributed to the impediment of shear band formation by the small grain size, facilitated by softer interfaces guiding early shear transformation zone (STZ) activities across the entire sample. Furthermore, a progressive reduction of elastic constants simulates the failure response observed in experiments. Our model predicts a critical grain size for the transition in agreement with molecular dynamics simulations and experiments, highlighting its potential for designing NGs with enhanced shear resistance. This mesoscale model enables the investigation of NG deformation with microstructural features on an experimentally-relevant spatial-temporal scale. This paves the way for tailoring NG microstructures to achieve enhanced mechanical performance, and opens new avenues for exploring the influence of interfaces in controlling shear localization.
Back to topN. Nakamura, K. Matsuura, A. Ishii,“In situ observation of morphological change of Pd-based bimetallic nanoparticles synthesized by co-sputtering”, Journal of Applied Physics, 134 (2023) 145301-1-6.

The formation process of Pd-based bimetallic nanoparticles synthesized by co-sputtering is investigated by performing in situ morphological observation using resistive spectroscopy. The segregation of the metal with lower surface energy on the nanoparticle surface is observed, and it is found that the formation process of the alloy nanoparticles tends to be similar to that of the nanoparticles composed of the core metal even when the atomic fraction of the shell metal is higher than that of the core metal. The co-sputtering process is simulated by the molecular dynamics analysis, and the observed formation process is theoretically confirmed.
Back to topNobutomo Nakamura, Koji Matsuura, Akio Ishii, and Hirotsugu Ogi,“Restructuring in bimetallic core-shell nanoparticles: Real-time observation”, Physical Review B, 105 (2022) 125401-1-5.
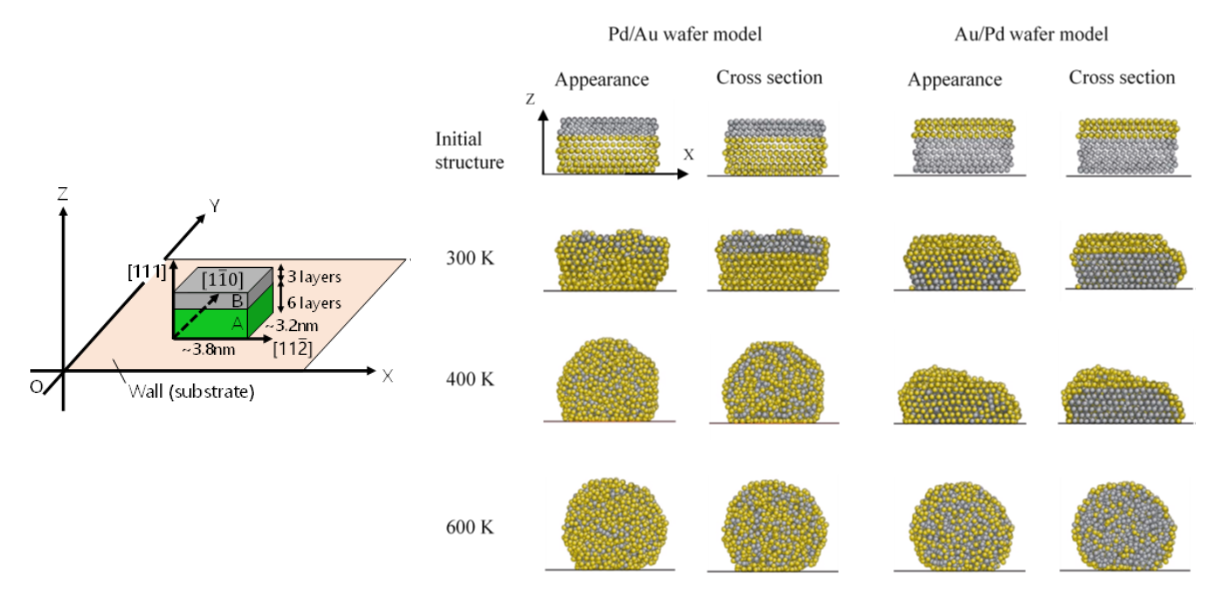
The formation process of core-shell bimetallic nanoparticles synthesized by sputtering onto a substrate is observed in real time using an originally developed acoustic technique. The technique enables us to evaluate the structural change of nanoparticles at room temperature without contacting the nanoparticles or substrate. In the experiments, the sputtering of metal A followed by metal B tended to form B-shell/A-core nanoparticles. However, in Pd-Au alloy system, notable restructuring occurred during synthesis, resulting in the formation of A-shell/B-core nanoparticles. The formation process is analyzed using the molecular dynamics simulation, revealing that this restructuring occurs on a short timescale, and high diffusivity of Au plays an important role.
Back to topPeijun Yu, Jun-Ping Du, Shuhei Shinzato, Fan-Shun Meng, Shigenobu Ogata,“Theory of history-dependent multi-layer generalized stacking fault energy— A modeling of the micro-substructure evolution kinetics in chemically ordered medium-entropy-alloys”, Acta Materialia, 224 (2022) 117504-1-12.
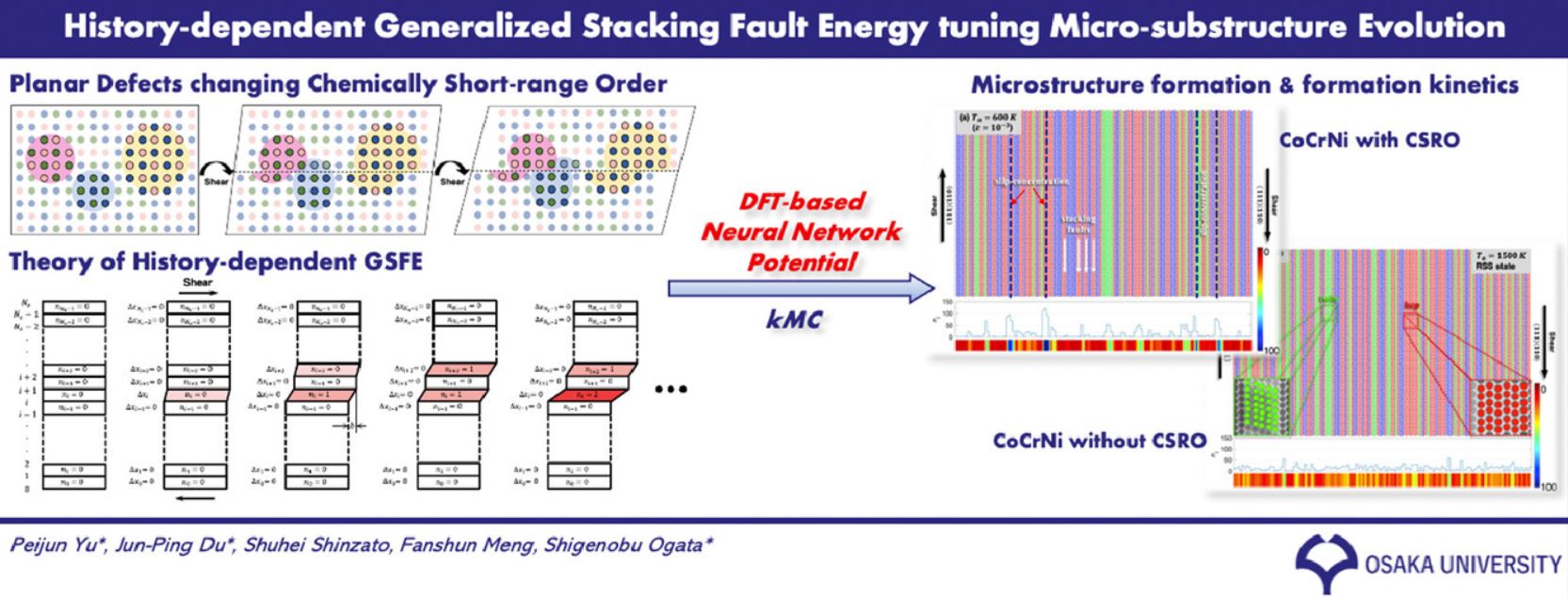
In this study, a chemical order related concept “history-dependent multi-layer generalized stacking fault energy” (HDML-GSFE) was proposed, and it was then demonstrated by employing the recent, very in- teresting multi-principal element alloy (CoCrNi medium-entropy alloys; MEA) with different chemical short-range order (CSRO) levels using a density functional theory (DFT)-based neural network interatomic potential. To demonstrate the impacts of the history dependency and interlayer (atomic interlayers of the slip system) coupling effect on the GSFE of CSRO MEAs, HDML-GSFEs were computed for different shear deformation pathways of the MEAs with different CSRO levels, such as interlayer multiple-time slipping, twin growth, and γ−ε(FCC-HCP) phase transformation. It was demonstrated that multiple-time slip- ping induces CSRO collapse, leading to local shear softening due to the history dependency of GSFE. In addition, it was found that the slipping of neighboring atomic interlayers is affected by the slipping re- sulting from the induced CSRO collapse of present interlayers because of the interlayer coupling effect of GSFE. Eventually, by employing a novel kinetic Monte Carlo (kMC) simulation method based on disloca- tion/disconnection loop nucleation events and using the HDML-GSFE with the history dependency and interlayer coupling effect, we proposed a laminated micro-substructure evolution that involves twinning and γ−εphase transformations subject to a finite shear strain rate and finite temperature.
Back to topXiang Wang, Sixue Zheng, Shuhei Shinzato, Zhengwu Fang, Yang He, Li Zhong, Chongmin Wang, Shigenobu Ogata, Scott X. Mao,“Atomistic processes of surface-diffusion-induced abnormal softening in nanoscale metallic crystals”, Nature Communications, 12 (2021) 5237-1-9.
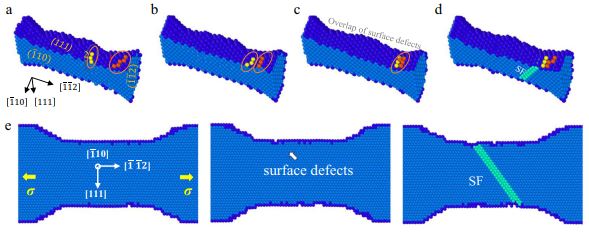
Ultrahigh surface-to-volume ratio in nanoscale materials, could dramatically facilitate mass transport, leading to surface-mediated diffusion similar to Coble-type creep in polycrystalline materials. Unfortunately, the Coble creep is just a conceptual model, and the associated physical mechanisms of mass transport have never been revealed at atomic scale. Akin to the ambiguities in Coble creep, atomic surface diffusion in nanoscale crystals remains largely unclear, especially when mediating yielding and plastic flow. Here, by using in situ nanomechanical testing under high-resolution transmission electron microscope, we find that the diffusion-assisted dislocation nucleation induces the transition from a normal to an inverse Hall-Petch-like relation of the strength-size dependence and the surface-creep leads to the abnormal softening in flow stress with the reduction in size of nanoscale silver, contrary to the classical “alternating dislocation starvation” behavior in nanoscale platinum. This work provides insights into the atomic-scale mechanisms of diffusion-mediated deformation in nanoscale materials, and impact on the design for ultrasmall-sized nanomechanical devices.
Back to topKazuma Ito, Hideaki Sawada, and Shigenobu Ogata, “Theoretical Prediction of Grain Boundary Segregation Using Nano-Polycrystalline Grain Boundary Model”, Materials Transactions, 62, 5, (2021) 575-581.[Translated paper. (Original Paper) Journal of Japan Institute of Metals and Materials, 84-7, (2020), 237-243.]
The importance of controlling grain boundary (GB) segregation is increasing, especially with the strengthening of steel nowadays. In this study, a theoretical prediction method for the amount of GB segregation for a solute element in polycrystals is established. This prediction method entails the development of a nano-polycrystalline GB model for simulating GBs in polycrystals, and the segregation energy of a solute element is comprehensively calculated for all atomic sites constituting the GB model by using an interatomic potential. From the obtained segregation energies, the segregation amount of the solute element at each atomic site is determined. Subsequently, each atomic site is classified based on its distance from the GB center and averaged to determine the segregation profile of the solute element for that distance from the GB center. By applying this method to the GB segregation of P in bcc-Fe and comparing the results with experimental findings, it is determined that this prediction method can deliver excellent prediction accuracies.
Back to topRuixiao Zheng, Jun-Ping Du, Si Gao, Hidetoshi Somekawa, Shigenobu Ogata, Nobuhiro Tsuji, “Transition of dominant deformation mode in bulk polycrystalline pure Mg by ultra-grain refinement down to sub-micrometer”, Acta Materialia, 198 (2020) 35-46.
Magnesium (Mg) and its alloys usually show relatively low strength and poor ductility at room temperature due to their anisotropic hexagonal close-packed (HCP) crystal structure that provides a limited number of independent slip systems. Here we report that unique combinations of strength and ductility can be realized in bulk polycrystalline pure Mg by tuning the predominant deformation mode. We succeeded in obtaining the fully recrystallized specimens of pure Mg having a wide range of average grain sizes, of which minimum grain size was 650 nm, and clarified mechanical properties and deformation mechanisms at room temperature systematically as a function of the grain size. Deformation twinning and basal slip governed plastic deformation in the conventional coarse-grained region, but twinning was suppressed when the grain size was refined down to several micro-meters. Eventually, grain boundary mediated plasticity, i.e., grain boundary sliding became dominant in the ultrafine-grained (UFG) specimen having a mean grain size smaller than 1 μm. The transition of the deformation modes led to a significant increase of tensile elongation and breakdown of Hall-Petch relationship. It was quantitatively confirmed by detailed microstructural observation and theoretical calculation that the change in strength and ductility arose from the distinct grain size dependence of the critical shear stress for activating different deformation modes.
Back to topKazuma Ito, Hideaki Sawada, Shigenobu Ogata,“Theoretical Prediction of Grain Boundary Segregation Using Nano-Polycrystalline Grain Boundary Model”, J. Japan Inst. Met. Mater. , Vol. 84, 7 (2020) 237-243.
Nucleation of transmutation products such as rhenium (Re) and osmium (Os) is a central issue contributing to changes in mechanical properties under neutron irradiation in fusion reactors. In particular, Re solutes in tungsten (W) not only affect hardening via radiation-induced precipitation but also have a notable softening effect. We explored the softening/strengthening behaviors of various solutes in a W matrix by density functional theory (DFT) calculations combined with a solid solution model. Our DFT calculations of the solutes show a clear trend in the interaction energy between different solutes and screw dislocations, which also influences the solid solution behavior. These predictions, based on a solid solution model and DFT calculations, reasonably reproduce the complex softening/strengthening behavior as a function of temperature and solute concentration. Notably, solutes such as Re have relatively weak attractive interactions and do not markedly influence the pinning effect; however, such solutes can reduce the energy barrier for kink pair nucleation. We conclude that this specific balance is the origin of macroscopic solid solution softening in dilute body-centered cubic alloys.
Back to topDe-Gang Xie, Zhi-Yu Nie, Shuhei Shinzato, Yue-Qing Yang, Feng-Xian Liu, Shigenobu Ogata, Ju Li, Evan Ma, Zhi-Wei Shan, "Controlled growth of single-crystalline metal nanowires via thermomigration across a nanoscale junction", Nature Communications, 10 (2019) 4478-1-8.
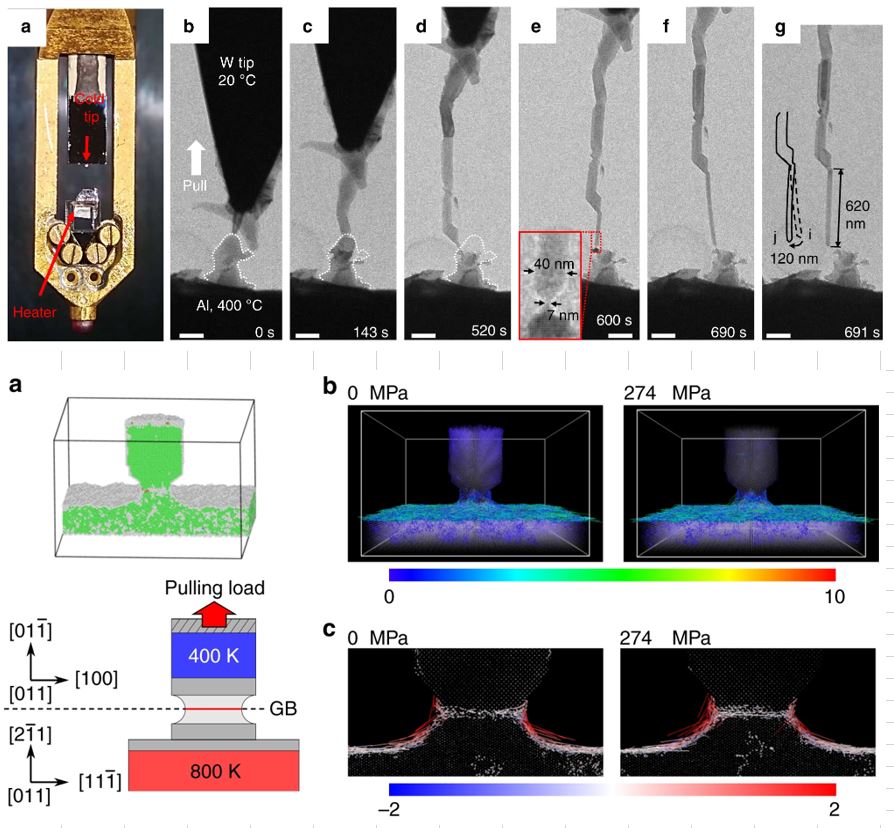
Mass transport driven by temperature gradient is commonly seen in fluids. However, here we demonstrate that when drawing a cold nano-tip off a hot solid substrate, thermomigration can be so rampant that it can be exploited for producing single-crystalline aluminum, copper, silver and tin nanowires. This demonstrates that in nanoscale objects, solids can mimic liquids in rapid morphological changes, by virtue of fast surface diffusion across short distances. During uniform growth, a thin neck-shaped ligament containing a grain boundary (GB) usually forms between the hot and the cold ends, sustaining an extremely high temperature gradient that should have driven even larger mass flux, if not counteracted by the relative sluggishness of plating into the GB and the resulting back stress. This GB-containing ligament is quite robust and can adapt to varying drawing directions and velocities, imparting good controllability to the nanowire growth in a manner akin to Czochralski crystal growth.
Back to topHiroshi Miyoshi, Hajime Kimizuka, Akio Ishii, Shigenobu Ogata, "Temperature-dependent nucleation kinetics of Guinier-Preston zones in Al-Cu alloys: An atomistic kinetic Monte Carlo and classical nucleation theory approach", Acta Materialia, 179 (2019) 262-272.
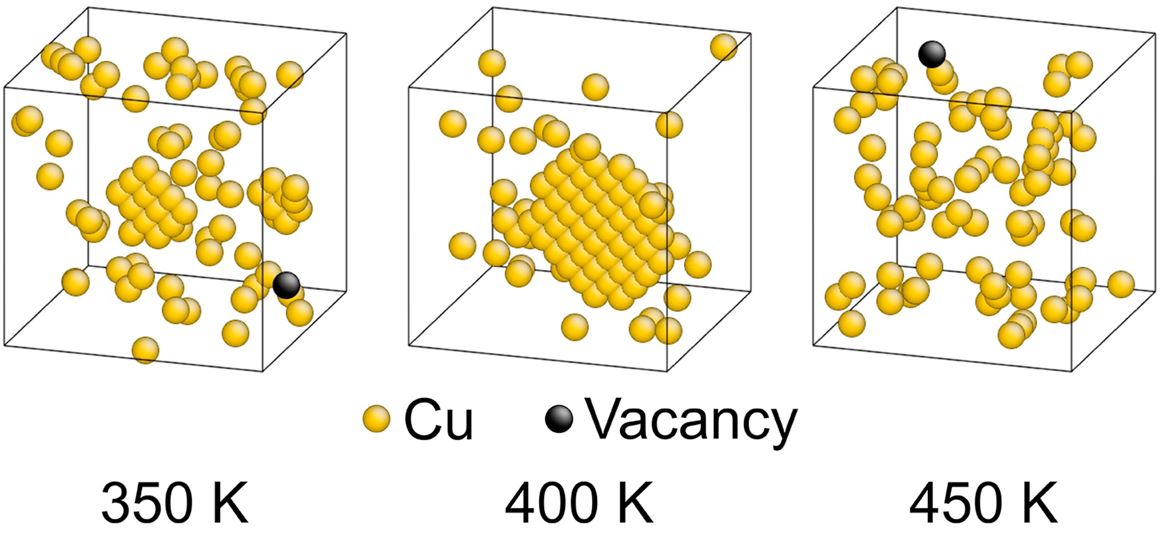
Guinier-Preston (GP) zones, which form in Al-Cu alloys, exhibit characteristic patterns of Cu-rich, disk-shaped atomic clusters along the {100} planes. GP zones have received much attention not only for their precipitation-hardening effect on the Al matrix, but also from fundamental interest in the physics and chemistry of the nanoscale organization of solute atoms. In this study, we established an atomistic kinetic Monte Carlo (kMC) modeling technique for exploring the nucleation and formation processes of GP zones in Al-Cu alloys by constructing an effective on-lattice multibody potential for a dilute Al-Cu-vacancy system by density functional theory calculations. Our model can describe the clustering of Cu atoms via successive exchanges with vacancies and reproduce the characteristic planar nanoprecipitates along the {100} planes in a manner consistent with the crystallographic nature of the GP zones in the early-stage aging of Al-Cu alloys. The time evolution and critical nucleus size of Cu clusters were characterized at various temperatures based on the kMC results. Consequently, we predicted the existence of an optimum temperature (i.e., nose temperature) for the formation of Cu clusters at which the cluster growth was maximized, which was attributable to the interplay between the critical nucleation barrier and the diffusion rate. In addition, the critical nucleus size and temperature for cluster formation were examined based on classical nucleation theory along with the developed multibody potential. These provided an insight into the competition between the enthalpic and entropic effects on the formation of GP zones in the Al-Cu system.
Back to topYuji Sato, Shuhei Shinzato, Takahito Ohmura, Shigenobu Ogata, "Atomistic prediction of the temperature- and loading-rate-dependent first pop-in load in nanoindentation", International Journal of Plasticity, 121 (2019) 280-292.
Nanoindentation has been used for a long time to investigate the mechanical properties of materials. A well-known displacement burst, the so-called “pop-in,” is usually observed during nanoindentation tests. In particular for the first pop-in has been well studied because it should be directly related to the fundamental mechanical strength of the local volume beneath the indenter. The first pop-in load measured for a defect-free local volume is corresponding to the onset load of homogeneous dislocation nucleation, while measured for a local volume with immobile defects is that of heterogeneous dislocation nucleation. The first pop-in is a thermally activated event under loading; thus, the pop-in load exhibits both temperature and loading-rate dependencies. Although theoretical studies have successfully explained the temperature and loading-rate dependencies, to the best of our knowledge, an atomistic prediction for specific materials which takes into account the stress state beneath the indenter has not yet been reported. In this study, we propose an atomistically informed multiscale (two-scale) modeling to predict the temperature and loading-rate dependencies of the first pop-in load for the homogeneous dislocation nucleation considering an atomistically estimated stress state beneath the indenter and stress-dependent activation energies of dislocation nucleation. Body-centered-cubic Fe and Ta are chosen as target metals, although the method is generally applicable to any ductile material. In addition to the homogeneous dislocation nucleation, we also developed a heterogeneous nucleation theory to explain the difference between the experimentally and atomistically obtained homogeneous nucleation pop-in loads.
Back to topGurcan Aral, Md Mahbubul Islam, Yun-Jiang Wang, Shigenobu Ogata and Adri C. T. van Duin, "Oxyhydroxide of metallic nanowires in a molecular H2O and H2O2 environment and their effects on mechanical properties" , Phys. Chem. Chem. Phys. 20 (2018) 17289.
To avoid unexpected environmental mechanical failure, there is a strong need to fully understand the details of the oxidation process and intrinsic mechanical properties of reactive metallic iron (Fe) nanowires (NWs) under various aqueous reactive environmental conditions. Herein, we employed ReaxFF reactive molecular dynamics (MD) simulations to elucidate the oxidation of Fe NWs exposed to molecular water (H2O) and hydrogen peroxide (H2O2) environment, and the influence of the oxide shell layer on the tensile mechanical deformation properties of Fe NWs. Our structural analysis shows that oxidation of Fe NWs occurs with the formation of different iron oxide and hydroxide phases in the aqueous molecular H2O and H2O2 oxidizing environments. We observe that the resulting microstructure due to pre-oxide shell layer formation reduces the mechanical stress via increasing the initial defect sites in the vicinity of the oxide region to facilitate the onset of plastic deformation during tensile loading. Specifically, the oxide layer of Fe NWs formed in the H2O2 environment has a relatively significant effect on the deterioration of the mechanical properties of Fe NWs. The weakening of the yield stress and Young modulus of H2O2 oxidized Fe NWs indicates the important role of local oxide microstructures on mechanical deformation properties of individual Fe NWs. Notably, deformation twinning is found as the primary mechanical plastic deformation mechanism of all Fe NWs, but it is initially observed at low strain and stress level for the oxidized Fe NWs.
Back to topShuhei Shinzato, Ei Kasuya, and Shigenobu Ogata, "Study of Interstitial Atom Diffusion in Nanowire using Duffusion Equation driven by Chemo-Mechanical Potential" , Journal of the Society of Materials Science, Japan, 67-2 (2018) 263-268.
Impurity atom distribution and its time evolution in cylindrical nanowire were studied using a diffusion equation based on chemo-mechanical potential, which considered coupling effects between chemical potential (chemical effect) and internal stress (mechanical effect) due to interstitial impurity atoms. We did consider diffusion blocking and nanowire yielding effects when we solved the diffusion equation. The yield criterion was described by comparing local volumetric strain caused by impurity atoms with critical volumetric strain at which the lattice instability appears. Obtained results used the diffusion equation with realistic materials parameters indicate a possibility of nanowire yielding due to interstitial atom diffusion and diffusion slowdown due to the yielding.
Back to topHajime Kimizuka, Shigenobu Ogata, and Motoyuki Shiga , “Mechanism of fast lattice diffusion of hydrogen in palladium: Interplay of quantum fluctuations and lattice strain”, Physical Review B, 97-1 (2018) 014102-1-11.
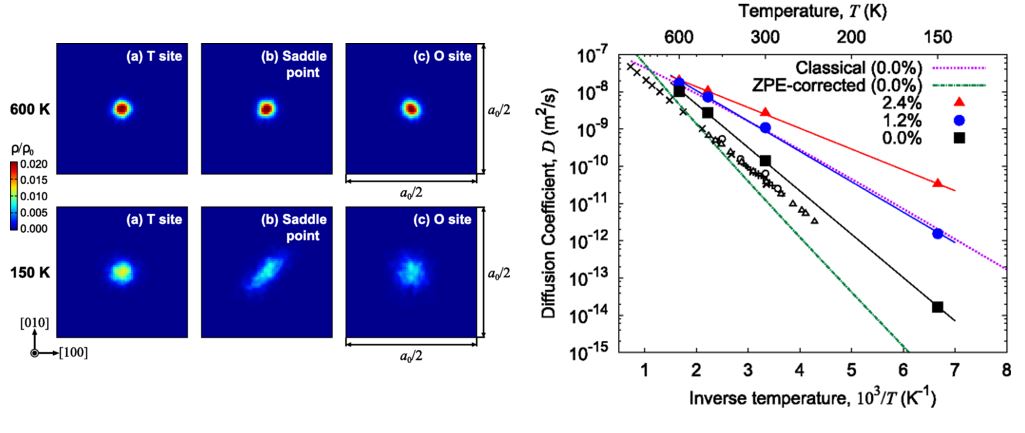
Understanding the underlying mechanism of the nanostructure-mediated high diffusivity of H in Pd is of recent scientific interest and also crucial for industrial applications. Here, we present a decisive scenario explaining the emergence of the fast lattice-diffusion mode of interstitial H in face-centered cubic Pd, based on the quantum mechanical natures of both electrons and nuclei under finite strains. Ab initio path-integral molecular dynamics was applied to predict the temperature- and strain-dependent free energy profiles for H migration in Pd over a temperature range of 150–600 K and under hydrostatic tensile strains of 0.0%–2.4%; such strain conditions are likely to occur in real systems, especially around the elastic fields induced by nanostructured defects. The simulated results revealed that, for preferential H location at octahedral sites, as in unstrained Pd, the activation barrier for H migration ( Q ) was drastically increased with decreasing temperature owing to nuclear quantum effects. In contrast, as tetrahedral sites increased in stability with lattice expansion, nuclear quantum effects became less prominent and ceased impeding H migration. This implies that the nature of the diffusion mechanism gradually changes from quantum- to classical-like as the strain is increased. For H atoms in Pd at the hydrostatic strain of ∼ 2.4 % , we determined that the mechanism promoted fast lattice diffusion ( Q = 0.11 eV) of approximately 20 times the rate of conventional H diffusion ( Q = 0.23 eV) in unstrained Pd at a room temperature of 300 K.
Back to topIchiro Kawarada, Ruixiao Zheng, Akinobu Shibata, Hidetoshi Somekawa, Shigenobu Ogata, and Nobuhiro Tsuji, “Mechanical Properties and Deformation Mechanism of Mg–Y Alloy with Various Grain Sizes”,Magnesium Technology 2017, (2017) 283-287.
In the present study, a Mg–Y dilute alloy was provided for a severe plastic deformation by high pressure torsion (HPT) and subsequent annealing. After the HPT by 5 rotations, nanocrystalline (NC) structures with an average grain size of 240 nm having deformed characteristics were obtained. Subsequent annealing at various temperatures for 2–60 min resulted in fully recrystallized structures with different average grain sizes ranging from 0.66 to 8.13 μm. Good balance of tensile strength and ductility could be realized in the fine grained specimens. For the specimen having a mean grain size of 2.13 μm, the yield strength and total tensile elongation were 180 MPa and 37%, respectively, which were much higher than those of pure Mg with a similar grain size. The significant contribution of Y on the microstructure and mechanical properties is discussed.
Back to topGurcan Aral, Yun-Jiang Wang, Shigenobu Ogata, and Adri C. T. van Duin, “Effects of oxidation on tensile deformation of iron nanowires: Insights from reactive molecular dynamics simulations”,Journal of Applied Physics, 120 (2016) 135104-1-15.
The influence of oxidation on the mechanical properties of nanostructured metals is rarely explored and remains poorly understood. To address this knowledge gap, in this work, we systematically investigate the mechanical properties and changes in the metallic iron (Fe) nanowires (NWs) under various atmospheric conditions of ambient dry O2 and in a vacuum. More specifically, we focus on the effect of oxide shell layer thickness over Fe NW surfaces at room temperature. We use molecular dynamics (MD) simulations with the variable charge ReaxFF force field potential model that dynamically handles charge variation among atoms as well as breaking and forming of the chemical bonds associated with the oxidation reaction. The ReaxFF potential model allows us to study large length scale mechanical atomistic deformation processes under the tensile strain deformation process, coupled with quantum mechanically accurate descriptions of chemical reactions. To study the influence of an oxide layer, three oxide shell layer thicknesses of ∼4.81 Å, ∼5.33 Å, and ∼6.57 Å are formed on the pure Fe NW free surfaces. It is observed that the increase in the oxide layer thickness on the Fe NW surface reduces both the yield stress and the critical strain. We further note that the tensile mechanical deformation behaviors of Fe NWs are dependent on the presence of surface oxidation, which lowers the onset of plastic deformation. Our MD simulations show that twinning is of significant importance in the mechanical behavior of the pure and oxide-coated Fe NWs; however, twin nucleation occurs at a lower strain level when Fe NWs are coated with thicker oxide layers. The increase in the oxide shell layer thickness also reduces the external stress required to initiate plastic deformation.
Back to topJun-Ping Du, Yun-Jiang Wang, Yu-Chieh Lo, Liang Wan, and Shigenobu Ogata, “Mechanism transition and strong temperature dependence of dislocation nucleation from grain boundaries: An accelerated molecular dynamics study”,Physical Review B, 94 (2016) 104110-1-8.
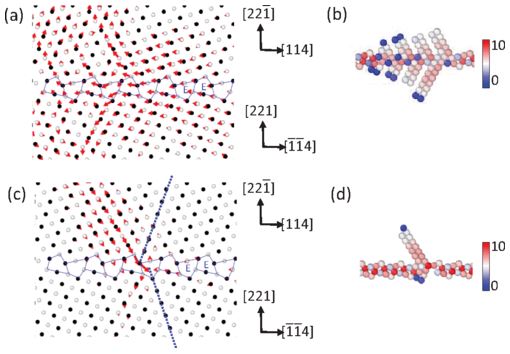
Accelerated molecular dynamics reveals a mechanism transition and strong temperature dependence of dislocation nucleation from grain boundaries (GBs) in Cu. At stress levels up to ~ 90% of the ideal dislocation nucleation stress, atomic shuffling at the E structural unit in a GB acts as a precursor to dislocation nucleation, and eventually a single dislocation is nucleated. At very high stress levels near the ideal dislocation nucleation stress, a multiple dislocation is collectively nucleated. In these processes, the activation free energy and activation volume depend strongly on temperature. The strain-rate dependence of the critical nucleation stress is studied and the result shows that the mechanism transition from the shuffling-assisted dislocation nucleation mechanism to the collective dislocation nucleation mechanism occurs during the strain rate increasing from 10^-4 s-1 to 10^10 s-1. The findings of the mechanism transition and strong temperature dependence of GB dislocation nucleation have been missed in the previous conventional molecular dynamics studies.
Back to topMing-Shuai Ding, Jun-Ping Du, Liang Wan, Shigenobu Ogata, Lin Tian, Evan Ma, Wei-Zhong Han, Ju Li, and Zhi-Wei Shan, “Radiation-Induced Helium Nanobubbles Enhance Ductility in Submicron-Sized Single-Crystalline Copper”, Nano Letters, 16-7 (2016) 4118-4124.
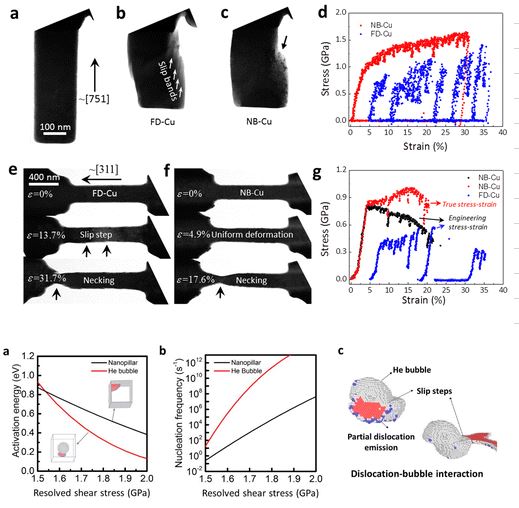
The workability and ductility of metals usually degrade with exposure to irradiation, hence the phrase “radiation damage”. Here, we found that helium (He) radiation can actually enhance the room-temperature deformability of submicron-sized copper. In particular, Cu single crystals with diameter of 100–300 nm and containing numerous pressurized sub-10 nm He bubbles become stronger, more stable in plastic flow and ductile in tension, compared to fully dense samples of the same dimensions that tend to display plastic instability (strain bursts). The sub-10 nm He bubbles are seen to be dislocation sources as well as shearable obstacles, which promote dislocation storage and reduce dislocation mean free path, thus contributing to more homogeneous and stable plasticity. Failure happens abruptly only after significant bubble coalescence. The current findings can be explained in light of Weibull statistics of failure and the beneficial effects of bubbles on plasticity. These results shed light on plasticity and damage developments in metals and could open new avenues for making mechanically robust nano- and microstructures by ion beam processing and He bubble engineering.
Back to topXu-Sheng Yang, Yun-Jiang Wang, Hui-Ru Zhai, Guo-Yong Wang, Yan-Jing Su, L. H. Dai, Shigenobu Ogata, and Tong-Yi Zhang, “Time-, stress-, and temperature-dependent deformation in nanostructured copper: Creep tests and simulations”, Journal of the Mechanics and Physics of Solids, 94 (2016) 191-206.
In the present work, we performed experiments, atomistic simulations, and high-resolution electron microscopy (HREM) to study the creep behaviors of the nanotwinned (nt) and nanograined (ng) copper at temperatures of 22 °C (RT), 40 °C, 50 °C, 60 °C, and 70 °C. The experimental data at various temperatures and different sustained stress levels provide sufficient information, which allows one to extract the deformation parameters reliably. The determined activation parameters and microscopic observations indicate transition of creep mechanisms with variation in stress level in the nt-Cu, i.e., from the Coble creep to the twin boundary (TB) migration and eventually to the perfect dislocation nucleation and activities. The experimental and simulation results imply that nanotwinning could be an effective approach to enhance the creep resistance of twin-free ng-Cu. The experimental creep results further verify the newly developed formula (Yang et al., 2016) that describes the time-, stress-, and temperature-dependent plastic deformation in polycrystalline copper.
Back to topKang Pyo Soa, Xiaohui Liua, Hideki Mori, Akihiro Kushima, Jong Gil Park, Hyoung Seop Kim, Shigenobu Ogata, Young Hee Lee, Ju Li, “Ton-scale metal–carbon nanotube composite: The mechanism of strengthening while retaining tensile ductility”, Extream Mechanics Letters, 8 (2016) 245-250.
One-dimensional carbon nanotubes (CNT), which are mechanically strong and flexible, enhance strength of the host metal matrix. However, the reduction of ductility is often a serious drawback. Here, we report significantly enhanced plastic flow strength, while preventing tensile ductility reduction, by uniformly dispersing CNTs in Al matrix. Nanoscale plasticity and rupturing processes near CNTs were observed by in-situ mechanical tests inside Transmission Electron Microscope (TEM). CNTs act like forest dislocations and have comparable density (∼10^14/m^2), and such 1D nano-dispersion hardening is studied in detail by in situ TEM and molecular dynamics simulations. Rupture-front blunting and branching are seen with in situ TEM, which corroborates the result from macro-scale tension tests that our Al+CNT nanocomposite is quite damage- and fault-tolerant. We propose a modified shear-lag model called “Taylor-dispersion” hardening model to highlight the dual roles of CNTs as load-bearing fillers and “forest dislocations” equivalent that harden the metal matrix, for the plastic strength of metal+CNT nanocomposite.
Back to topHiroshi Okuda, Michiaki Yamasaki, Yoshihito Kawamura, Masao Tabuchi, and Hajime Kimizuka, “Nanoclusters first: a hierarchical phase transformation in a novel Mg alloy”, Scientific Reports, 5 (2015) 14186-1-6.
The Mg-Y-Zn ternary alloy system contains a series of novel structures known as long-period stacking ordered (LPSO) structures. The formation process and its key concept from a viewpoint of phase transition are not yet clear. The current study reveals that the phase transformation process is not a traditional spinodal decomposition or structural transformation but, rather a novel hierarchical phase transformation. In this transformation, clustering occurs first, and the spatial rearrangement of the clusters induce a secondary phase transformation that eventually lead to two-dimensional ordering of the clusters. The formation process was examined using in situ synchrotron radiation smallangle X-ray scattering (SAXS). Rapid quenching from liquid alloy into thin ribbons yielded strongly supersaturated amorphous samples. The samples were heated at a constant rate of 10 K/min. and the scattering patterns were acquired. The SAXS analysis indicated that small clusters grew to sizes of 0.2 nm after they crystallized. The clusters distributed randomly in space grew and eventually transformed into a microstructure with two well-defined cluster-cluster distances, one for the segregation periodicity of LPSO and the other for the in-plane ordering in segregated layer. This transformation into the LPSO structure concomitantly introduces the periodical stacking fault required for the 18R structures.
Back to topMarco Fronzi, Hajime Kimizuka, and Shigenobu Ogata, “Atomistic investigation of the vacancy-assisted diffusion mechanism in Mg ternary (Mg-RE-M) alloys”, Computational Materials Science, 98 (2015), 76-82.
To investigate the kinetics of the formation of solute cluster structures in some of the Mg ternary alloys, we perform a first-principles analysis of some fundamental quantities in doped Mg lattices. We calculate interaction energies between vacancy and solute atoms in both Mg hexagonal close packed (HCP) and face-centered cubic (FCC) crystal structures. In particular, we consider in this work Al, Gd, Y, and Zn solute atoms. Also, to understand the diffusion mechanism, we calculate vacancy-assisted diffusion for Mg and solute atoms in HCP and FCC lattices using the nudge elastic band method.
Back to topWei-Zhong Han, Ling Huang, Shigenobu Ogata, Hajime Kimizuka, Zhao-Chun Yang, Christopher Weinberger, Qing-Jie Li, Bo-Yu Liu, Xi-Xiang Zhang, Ju Li, Evan Ma, Zhi-Wei Shan, “From “smaller is stronger” to “size-independent strength plateau”: towards measuring the ideal strength of iron”, Advanced Materials, 27 (2015) 3385-3390.
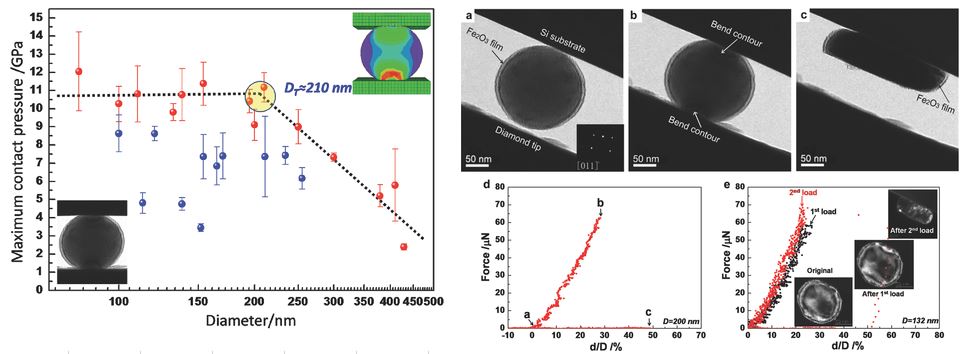
The trend from “smaller is stronger” to “size-independent strength plateau” is observed in the compression of spherical iron nanoparticles. When the diameter of iron nanospheres is less than a critical value, the maximum contact pressure saturates at 10.7 GPa, corresponding to a local shear stress of ≈9.4 GPa, which is comparable to the theoretical shear strength of iron.
Back to topHajime Kimizuka, Shu Kurokawa, Akihiro Yamaguchi, Akira Sakai, and Shigeobu Ogata, “Two-Dimensional Ordering of Solute Nanoclusters at a Close-Packed Stacking Fault: Modeling and Experimental Analysis”, Scientific Reports, 4 (2014), 7318-1-7318-9.
Predicting the equilibrium ordered structures at internal interfaces, especially in the case of nanometer-scale chemical heterogeneities, is an ongoing challenge in materials science. In this study, we established an ab-initio coarse-grained modeling technique for describing the phase-like behavior of a close-packed stacking-fault-type interface containing solute nanoclusters, which undergo a two-dimensional disorder-order transition, depending on the temperature and composition. Notably, this approach can predict the two-dimensional medium-range ordering in the nanocluster arrays realized in Mg-based alloys, in a manner consistent with scanning tunneling microscopy-based measurements. We predicted that the repulsively interacting solute-cluster system undergoes a continuous evolution into a highly ordered densely packed morphology while maintaining a high degree of six-fold orientational order, which is attributable mainly to an entropic effect. The uncovered interaction-dependent ordering properties may be useful for the design of nanostructured materials utilizing the self-organization of two-dimensional nanocluster arrays in the close-packed interfaces.
Back to topHajime Kimizuka and Shigenobu Ogata, "Predicting Atomic Arrangement of Solute Clusters in Dilute Mg Alloys", Materials Research Letters, 1-4 (2013) 213-219.
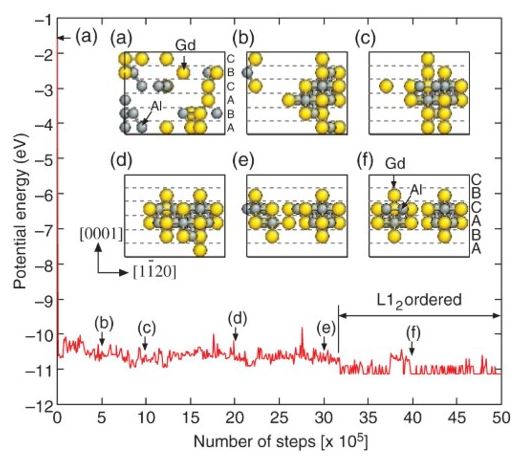
Theoretical prediction of the heterogeneous atomic structures in multicomponent alloys is one of the most challenging issues in materials science. Here we present a first-principles demonstration of this by constructing an on-lattice effective multibody potential model to describe the energetics of hexagonal close-packed Mg crystals containing stacking faults (SFs) with Al and Gd substitutions. Remarkably, we showed that our intuitive model can describe the segregation of solute atoms to SFs and reproduce the characteristic short-range chemical order in dilute Mg-Gd and Mg-Al-Gd systems, including long-period stacking ordered structures, in a manner consistent with recent scanning transmission electron microscopy measurements.
Back to topYun-Jinag Wang, Guo-Jie Jason Gao, and Shigenobu Ogata, "Atomistic understanding of diffusion kinetics in nanocrystals from molecular dynamics simulations", Physical Review B, 88-11 (2013) 115413-17.
Understanding the grain size effect on diffusion in nanocrystals has been hampered by the difficulty of measuring diffusion directly in experiments. Here large-scale atomistic modeling is applied to understand the diffusion kinetics in nanocrystals. Enhanced short-circuit diffusivity is revealed to be controlled by the rule of mixtures for grain-boundary diffusion and lattice diffusion, which can be accurately described by the Maxwell-Garnett equation instead of the commonly thought Hart equation, and the thermodynamics of pure grain-boundary self-diffusion is not remarkably affected by varying grain size. Experimentally comparable Arrhenius parameters with atomic detail validate our results. We also propose a free-volume diffusion mechanism considering negative activation entropy and small activation volume. These help provide a fundamental understanding of how the activation parameters depend on size and the structure-property relationship of nanostructured materials from a physical viewpoint.
Back to topShin Yamamoto, Yun-Jiang Wang, Akio Ishii, and Shigenobu Ogata, "Atomistic Design of High Strength Crystalline-Amorphous Nanocomposites", Materials Transactions, 54-9 (2013) 1592-1596.
There is a long-standing demand for materials which could simultaneously demonstrate multiple promising properties like high strength, good ductility and toughness. In this study, a three-dimensional bulk nanocomposite material which is composed of nanoscale crystalline metal and metallic glass is revealed to present high strength and potentially good ductility by molecular dynamics. A critical high strength is achieved by varying the ratio between crystalline and amorphous phase. The critical strength is revealed to be higher than that expected from the rule of mixture. The mechanism underlying the occurrence of critical strength in the nanocomposite is elucidated by the interaction between dislocation and matrix of amorphous phase. Our concept could guide the engineers to design more advanced bulk nanostructured materials.
Back to topHajime Kimizuka, Marco Fronzi, and Shigenobu Ogata, "Effect of alloying elements on in-plane ordering/disordering of solute clusters in Mg-based long-period stacking ordered structures: A first-principles analysis", Scripta Materialia, 69-8 (2013) 594-597.
Using density functional theory, we characterized the in-plane binding between L12-type solute clusters in Mg-M-RE (M = Al, Zn; RE = Y, Gd) long-period stacking ordered (LPSO) structures. The difference between the Al and Zn concentrations within the clusters determines whether the intercluster interaction is attractive or repulsive. Incomplete in-plane ordering observed experimentally in Mg-Zn-Y LPSO structures was suggested to be caused by the unlinked nature of the clusters owing to their significant inward contraction.
Back to topGuo-Jie J.Gao, Yun-Jiang Wang, and Shigenobu Ogata, "Studying the elastic properties of nanocrystalline copper using a model of randomly packed uniform grains", Computational Materials Science, 79 (2013) 56-62.
We develop a new Voronoi protocol, which is a space tessellation method, to generate a fully dense (containing no voids) model of nanocrystalline copper with precise grain size control; we also perform uniaxial tensile tests using molecular dynamical (MD) simulations to measure the elastic moduli of the grain boundary and the grain interior components at 300 K. We find that the grain boundary deforms more locally compared with the grain core region under thermal vibrations and is elastically less stiff than the core component at finite temperature. The elastic modulus of the grain boundary is lower than 30% of that of the grain interior. Our results will aid in the development of more accurate continuum models of nanocrystalline metals.
Back to topYun-jiang Wang, Akio Ishii, and Shigenobu Ogata, "Entropic effect on creep in nanocrystalline metals", Acta Materialia, 61-10 (2013) 3866-3871.
We report a significant entropic effect on creep of nanocrystalline metal using molecular dynamics. Our simulations reveal that the activation entropy may contribute a multiplicative factor of many orders of magnitude to the steady-state creep rate. The relationship between activation entropy and enthalpy obeys an empirical Meyer-Neldel compensation rule. The activation volume is found to decrease with increasing temperature for dislocation nucleation creep, which agrees well with experimental results. The study opens up an avenue for quantitatively discussing the entropic effects on various thermally activated deformations in nanocrystals.
Back to topAkio Ishii, Ju Li, and Shigenobu Ogata, "”Conjugate Channeling” Effect in Dislocation Core Diffusion: Carbon Transport in Dislocated BCC Iron", PloS one, 8-4 (2013) e60586-1-7.
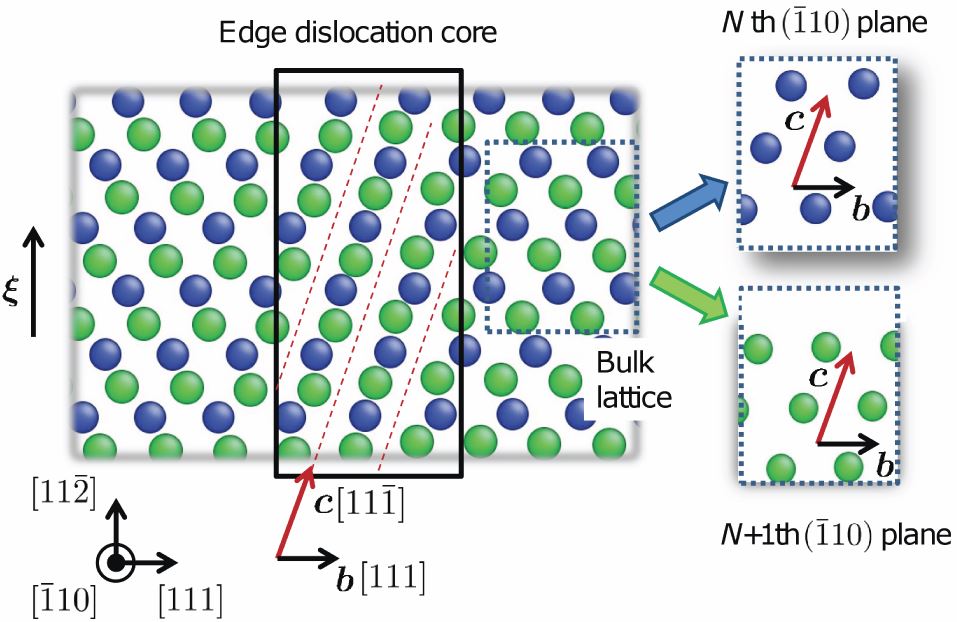
Dislocation pipe diffusion seems to be a well-established phenomenon. Here we demonstrate an unexpected effect, that the migration of interstitials such as carbon in iron may be accelerated not in the dislocation line direction ξ, but in a conjugate diffusion direction. This accelerated random walk arises from a simple crystallographic channeling effect. c is a function of the Burgers vector b, but not ξ, thus a dislocation loop possesses the same everywhere. Using molecular dynamics and accelerated dynamics simulations, we further show that such dislocation-core-coupled carbon diffusion in iron has temperature-dependent activation enthalpy like a fragile glass. The 71° mixed dislocation is the only case in which we see straightforward pipe diffusion that does not depend on dislocation mobility.
Back to topYun-Jiang Wang, Guo-Jie Jason Gao, and Shigenobu Ogata, "Size-dependent transition of deformation mechanism, and nonlinear elasticity in Ni3Al nanowires", Applied Physics Letters, 102 (2013) 041902-1-5.
A size-dependent transition of deformation mechanism is revealed in Ni3Al nanowire under atomistic uniaxial tension. Deformation twinning is replaced by phase transformation when the diameter of Ni3Al nanowire reduces to a critical value near 4 nm. Enhanced size-dependent nonlinear elasticity is observed in the nanowires, in comparison to their bulk counterpart which is benchmarked by combined density functional and atomistic study. This study provide fundamental understanding on the size-dependent deformation mechanisms of nanostructured alloys.
Back to topYun-jiang Wang, Akio Ishii, and Shigenobu Ogata, "Grain Size Dependence of Creep in Nanocrystalline Copper by Molecular Dynamics", Materials Transactions, 53-1 (2012) 156-160.
The grain size dependence of creep is critical to understand the plastic deformation mechanism of nanoscale metals. Here we used molecular dynamics to study the stress-induced grain size exponent transition in creep of nanocrystalline copper. The grain size exponent was found to initially increase with increasing stress, then decrease after some critical stress. The derived grain size exponents are in agreement with experimental results for diffusional and grain boundary sliding creep at low stress. While, the founded decreasing grain size exponent with increasing stress for dislocation nucleation creep in nanocrystal is in contrast with conventional materials. We propose a constitutive equation for dislocation nucleation governed creep in nanocrystal to explain its grain size dependence transition with stress.
Back to topYun-Jiang Wang, Akio Ishii, and Shigenobu Ogata, "Transition of creep mechanism in nanocrystalline metals", Physical Review B, 84-22 (2011) 224102-1-7.
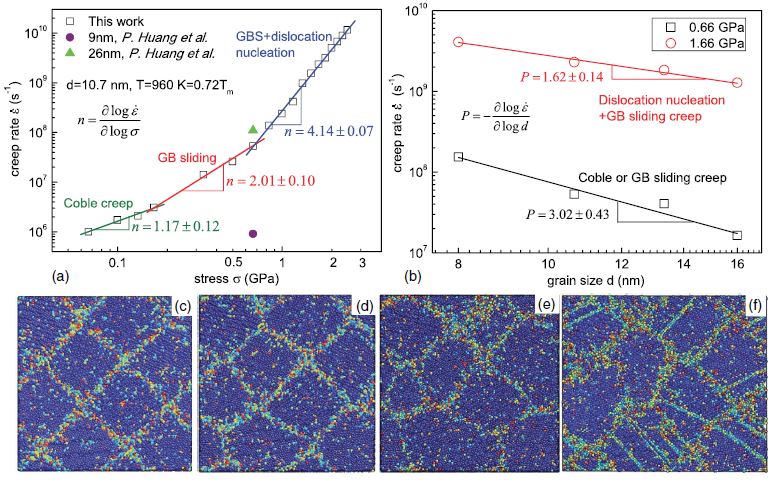
Understanding creep mechanisms with atomistic details is of great importance to achieve the mechanical and thermodynamical stabilities of nanocrystalline (NC) metals over a wide temperature range. Here we report a molecular dynamics analysis of creep in NC copper dominated by competing deformation mechanisms. We found the dominating creep mechanism transits from grain boundary (GB) diffusion to GB sliding, and then dislocation nucleation with increasing stress. The derived stress exponent, small activation volume of 0.1−10b3, and grain size exponent all agree quantitatively with experimental values. We proposed a stress-temperature deformation map in NC metals accommodated by the competition among different stress-driven, thermally activated processes. The model is general to answer the question why deformation mechanism transits with stress in NC metals.
Back to topFanyan Meng, Guisheng Wang, Sanqiang Shi, and Shigenobu Ogata, "A streched carbon nanotube with a high-density of topological defect", Advanced Materials Research, 236-238 (2011) 2225-2228.
We have developed a theoretical method to obtain a single-walled carbon nanotube (SWCNT) with a high density of topological defects. Carbon nanotubes (CNTs) sustain elastic elongation up to 15-30% at low temperature because of the sufficiently high barrier of bond rotations. A large number of topological defects are activated simultaneously and widely distributed over the entire tube wall after heating the stretched tube to an elevated temperature. This is driven by the internal energy of the strained carbon nanotubes. The manner in which topological defects are distributed is affected by the initial strain and the heating temperature. Nanotubes with a large number of topological defects achieve the elongation without breaking.
Back to topRyusuke Nakamura, Manabu Ishimaru, Akihiko Hirata, Kazuhisa Sato, Masakazu Tane, Hajime Kimizuka, Takehiko Shudo, Toyohiko Konno, Hideo Nakajima, "Enhancement of nanovoid formation in annealed amorphous Al2O3 including W", Journal of Applied Physics, 110-6 (2011) 064324-1-7.
The effect of W on the nanovoid formation in annealed amorphous Al2O3 was studied by transmission electron microscopy and molecular dynamics simulations. A comparison of the void formation behavior in electron-beam deposited Al2O3 (without W) and resistance-heating deposited Al2O3 (with 10 at. % W) revealed that W enhances the formation and growth of nanovoids. An analysis of the pair distribution function (PDF) in both types of amorphous Al2O3 showed that the introduction of W into amorphous Al2O3 brings about a significant change in the amorphous structure. Furthermore, it was found by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) that sub-nm sized W clusters exist in as-deposited Al2O3 prepared by resistance-heating and then dissolve in the amorphous matrix with annealing. The combination of PDF analysis and HAADF-STEM observation provides evidence that the enhancement of void formation originates in the heterogeneous short-range atomic configurations induced by the addition of W.
Back to topHajime Kimizuka and Shigenobu Ogata, "Slow diffusion of hydrogen at a screw dislocation core in alpha-iron", Physical Review B, 84-2 (2011) 024116.
Here we demonstrate and characterize the H-diffusion behavior around a screw dislocation in body-centered cubic (bcc) α-Fe by performing path-integral molecular dynamics modeling and adopting an ab initio–based potential. Counterintuitively, our results indicate that the H diffusivity along the dislocation line is significantly lower than lattice diffusion. Thus, the “fast” pipe diffusion does not occur for H in α-Fe.
Back to topHajime Kimizuka, Hideki Mori, and Shigenobu Ogata, "Effect of Temperature on Fast Hydrogen Diffusion in Iron: A Path-Integral Quantum Dynamics Approach", Physical Review B, 83-9 (2011) 094110-1-7.
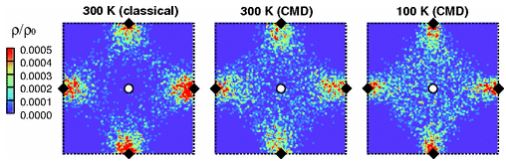
Here we explicitly present the diffusion coefficients (D) and activation energies (Ea) of interstitial H in α-Fe over a temperature range of 100 to 1000 K. These values were predicted by applying path-integral molecular dynamics modeling based on first principles. The obtained D and Ea values exhibit clear non-Arrhenius temperature dependence and a transition from quantum to classical behavior at around 500 K. Our results show that the quantum effects not only significantly lower the diffusion barrier but also change the diffusion pathway even at room temperature; thus, fast diffusion becomes possible.
Back to topHideki Mori, Hajime Kimizuka, and Shigenobu Ogata, "Microscopic Phase-Field Modeling of Edge and Screw Dislocation Core Structures and Peierls Stresses of BCC Iron", Journal of the Japan Institute of Metals, 75-2 (2011) 104-109. [in Japanese].
We investigate edge and screw dislocation core structures and Peierls stresses of BCC iron using microscopic phase-field (MPF) modeling. Parameters needed for the MPF modeling, such as the generalized-stacking-fault (GSF) energy, are determined based on first principles density functional theory (DFT) calculations. Screw dislocation core has six-fold symmetric structure and 0.05 nm width. The edge dislocation core is three times wider than the screw dislocation core. The Peierls stresses of edge and screw dislocations are estimated as 0.07 GPa and 2.8 GPa, respectively.
Back to topMasahiro Yamamoto, Kohei Kunizawa, Akinori Fujinami, Shigenobu Ogata, and Yoji Shibutani, "Formation of Atomistic Island in Al Film Growth by Kinetic Monte Carlo", Journal of Computational Science and Technology, 3-1 (2009) 148-158.
Kinetic Monte Carlo (KMC) method realizes the millisecond or second order atomistic thin film growth. Twenty five kinds of events which may occur on Al(111) surface were classified. An attempt frequency and an activation energy of each event were defined using vibration analyses and nudged elastic band (NEB) method by which the minimum energy path (MEP) can be reasonably predicted. Temperature and deposition rate dependences of Al(111) film growth were intensively investigated in the present paper. The higher temperature and the lower rate drive the layer-by-layer film structural change. Two types of islands (fcc and hcp) were seen by modeling without considering the events of diffusion of dimer and trimer, while only fcc islands remain with considering such events. Thus, we find that the primitive events of diffusion of dimer and trimer take important roles in determination of surface morphology.
Back to topShigenobu Ogata, Yoshitaka Umeno, and Masanori Kohyama, "First-Principles Approaches to Intrinsic Strength and Deformation of Materials: Perfect Crystals, Nano-Structures, Surfaces and Interfaces", Modelling and Simulation in Materials Science and Engineering, 17-1 (2009) 013001-1-33.
First-principles studies on the intrinsic mechanical properties of various materials and systems through ab initio tensile and shear testing simulations based on density-functional theory are reviewed. For various materials, ideal tensile and shear strength and features of the deformation of bulk crystals without any defects have been examined, and the relation with the bonding nature has been analyzed. The surfaces or low-dimensional nano-structures reveal peculiar strength and deformation behavior due to local different bonding nature. For grain boundaries and metal/ceramic interfaces, tensile and shear behaviors depend on the interface bonding, which impacts on the research of real engineering materials. Remaining problems and future directions in this research field are discussed.
Back to topAkinori Fujinami, Shigenobu Ogata, Hajime Kimizuka, and Yoji Shibutani, "The Energetics of Large Deformations of a Single Polyimide Molecular Chain: DFT and MO Calculations", Macromolecular Theory & Simulations, 17-9 (2008) 488-495.
The large-deformation energetics of a single molecular chain of the rod-like polyimide PMDA-PDA was investigated using DFT, ab initio MO and semi-empirical MO methods. The force/displacement curves were calculated from tensile testing simulations along the axis of the molecular chain, allowing a discussion of the distribution and change of local strain of the molecular chain. The deformation behavior of a single PMDA-PDA molecular chain under finite deformations as functions of bending angle and dihedral angle between PMDA and PDA groups are compared. It is found that the semi-empirical MO calculations provide sufficient accuracy to express the energetics of large deformations except for compressive deformation.
Back to topKohei Kunizawa, Masahiro Yamamoto, Shigenobu Ogata, and Yoji Shibutani, "Step-Growth Anisotropy on Thin Film Epitaxial Process", Journal of the Society of Materials Science, Japan, 57-8 (2008) 780-785.
Anisotropic growth process of two kinds of steps on Al(111) substrates is performed using kinetic Monte Carlo (kMC) method. Employed kMC parameters of activation energy and attempt frequency are estimated by nudged elastic band (NEB) method and transition state theory. Obtained set of results suggest that degree of the anisotropic growth clearly depends on substrate temperature and deposition rate. We find microscopic origin of the anisotropic growth is difference of diffusion rates along {111} and {100} steps, and there is a particular growth condition in which strong anisotropy is observed. At high deposition rate and low temperature, new islands which are easily generated on terraces, hinder the growth anisotropy weaker.
Back to topHajime Kimizuka, Shigenobu Ogata, and Ju Li, "Hydrostatic Compression and High-Pressure Elastic Constants of Coesite, SiO2", Jounal of Applied Physics, 103-5 (2008) 053506-1-4.
Using density-functional theory, we computed all the independent elastic constants of coesite, a high-pressure polymorph of silica, as functions of pressure up to 15 GPa. The results are in good agreement with experimental measurements under ambient conditions. Also, the predicted pressure-dependent elastic properties are consistent with x-ray data in the literature concerning lattice strains at high pressures. We find that coesite, like quartz, exhibits a gradual softening of a shear modulus B44 with increasing pressure, in contrast to the rising bulk modulus.
Back to topMasahiro Yamamoto, Akinori Fujinami, Shigenobu Ogata, and Yoji Shibutani, "Hybridized Atomistic Modeling of Migration Observed on Thin Film Surface by Incident Particles", Journal of Computational Science and Technology, 1-1 (2008) 14-21.
Innovative thin film technology to realize the finer electric devices needs to understand the atomic level process of film growth and its relationship to the film characterization. In this paper, the long film growth phenomena for a few micro-second order with the short severe collisions by incident particles are analyzed by the proposed hybridized atomistic modeling. This method combined molecular dynamics (MD) with kinetic Monte Carlo (KMC) can directly treat two types of events of deposition and diffusion, which have quite different time scales. The solutions suggest that the large incident kinetic energy of deposited atoms compatible to the realistic physical vapor deposition (PVD) impels to fluctuate the equilibrium on Al (111) surface very drastically and affects the atomic level surface morphology. It is found that the faster incident atoms with 1.0×104 m/s can make the smoother surface than those with the velocity of 1.0×103 m/s. This is due to much activated atomic migration, which can be realized only by MD.
Back to topHideki Mori, Shigenobu Ogata, Ju Li, Seiji Akita, and Yoshikazu Nakayama, "Plastic Bending and Shape Memory Effect of Double-Wall Carbon Nanotubes", Physical Review B, 76-16 (2007) 165405.
Plastic bending of (5,5)@(10,10) double-wall carbon nanotube is analyzed using nudged elastic band minimum energy path calculations. At lower applied bending curvature, only the outer tube deforms plastically. However, at higher bending curvature, both the inner and outer tubes deform plastically. We find that the plastic deformation of the outer tube is more difficult than that of isolated single-wall carbon nanotube of the same diameter due to tube-tube interactions. In contrast, the plastic deformation of the inner tube is not strongly affected by the presence of the outer tube. We also analyze the shape-memory effect (SME) discovered experimentally, which is a thermal recovery process from the plastically bent state to the straight defect-free state, which can be repeated multiple times. We analyze the physics behind SME of carbon nanotubes, which is quite different from that of traditional shape-memory alloys.
Back to topFanyan Meng, Shigenobu Ogata, Dongsheng Xu, Yoji Shibutani, and S. Q. Shi, "Thermal conductivity of an ultrathin carbon nanotube with an X-shaped junction", Physical Review B, 75-20 (2007), 205403-1-6.
The thermal conductivity of the ultrathin carbon nanotube with and without an X-shaped junction was investigated using nonequilibrium molecular-dynamics simulations. The ultrathin carbon nanotube exhibits superhigh thermal conductivity. The thermal conductivity of the nanotube with junctions was 20–80% less than that of a straight nanotube depending on temperature. There is a jump in the temperature profile around the junction, contributing to a larger temperature gradient and reduction in the thermal conductivity. The thermal conductivity of armchair nanotube junctions is sensitive to the topological structures at the junction region.
Back to topMasahiro Yamamoto, Kohei Kunizawa, Akinori Fujinami, Shigenobu Ogata, and Yoji Shibutani, "Formation of Atomistic Island in Al Film Growth by Kinetic Monte Carlo", Transactions of the JSME A, 73-728 (2007) 490-497. [in Japanese]
Kinetic Monte Carlo (KMC) method realizes the millisecond or second order atomistic thin film growth. Twenty five kinds of events which may occur on Al (111) surface were classified. An attempt frequency and an activation energy of each event were defined using vibration analyses and nudged elastic band (NEB) method by which the minimum energy path (MEP) can be reasonably predicted. Temperature and deposition rate dependences of Al(lll) film growth were intensively investigated in the present paper. The higher temperature and the lower rate drive the layer-by-layer film structural change. Two types of islands (fee and hep) were seen by modeling without considering the events of diffusion of dimer and trimer, while only fee islands remain with considering such events. Thus, we find that the primitive events of diffusion of dimer and trimer take important roles in determination of surface morphology.
Back to top